摘要:生物質作為一種綠色可再生資源,具有替代化石資源以生產(chǎn)高價值化學品和生物燃料的潛力。由于生物質原料中含氧量較高,因此加氫脫氧(HDO)反應成為其轉化過程中的關鍵步驟。氫碘酸(HI)由
生物質作為一種綠色可再生資源,具有替代化石資源以生產(chǎn)高價值化學品和生物燃料的潛力。由于生物質原料中含氧量較高,因此加氫脫氧(HDO)反應成為其轉化過程中的關鍵步驟。氫碘酸(HI)由于其強親核性和還原性,能夠在溫和條件下有效催化生物質的氫解,并且在某些生物質轉化過程中,可實現(xiàn)無金屬催化劑的應用。本文綜述了“HI-金屬催化劑-H?”催化體系及“HI-氫供體”無金屬催化體系在生物質HDO反應中的研究進展,今后對HI體系的探究應當關注機理研究,擴大HI在生物質定向轉化的利用范圍,重視反應過程對環(huán)境的影響。
關鍵詞: 生物質;加氫脫氧;氫碘酸
論文《氫碘酸催化在生物質加氫脫氧過程中的應用》發(fā)表在《中國科學:化學》,版權歸《中國科學:化學》所有。本文來自網(wǎng)絡平臺,僅供參考。
1 引言
隨著化石能源儲量的逐漸枯竭及公眾對環(huán)境問題的不斷關注,發(fā)展可再生資源以替代傳統(tǒng)化石資源(如石油、煤和天然氣)用于生產(chǎn)高附加值的化學品、生物燃料和功能材料,不僅能夠推動社會的可持續(xù)發(fā)展,還將助力我國實現(xiàn)節(jié)能減排和環(huán)境保護的目標[1,2]。生物質作為一種綠色且可再生的資源,理論上可通過自然界的光合作用實現(xiàn)碳中和的能量循環(huán),因而具有部分替代化石資源提供燃料和化學品的巨大潛力[3]。其中,木質纖維素是最為豐富的生物質形式,主要由纖維素(占比40%~50%)、半纖維素(占比20%~30%)及木質素(占比10%~35%)構成[4]。半纖維素與纖維素分別是由五碳糖和六碳糖單體組成的多聚體,而木質素是由苯基丙烷單元構成的三維無定型芳香族有機聚合物。
生物質原料中往往含有大量的氧元素(通常占干物質總量40%~50%),這導致它們的能量密度低于傳統(tǒng)的化石燃料,通過加氫脫氧反應對其進行處理可以降低產(chǎn)物中的氧含量,從而增加其能量密度,進而將其轉化為化學品和高密度生物燃料。然而,生物質加氫脫氧反應通常需要較高的反應溫度和貴金屬催化劑,且其選擇性也有待進一步提高[5~8]。
碘(I?)是合成各類無機及有機碘化合物的重要原料,同時也是人和動植物體必需的微量化學元素[9]。在鹵素中,I?具有最大的原子半徑,這使得C–I鍵長度最長且鍵能最低,因此也最易于斷裂。I?在鹵族元素中的還原電位最低(I?: +0.54 V;F?: +2.87 V;Cl?: +1.36 V;Br?: +1.07 V),這表明碘(I?)與碘離子(I?)之間相互轉化的熱力學可能性更高[10]。I?的共軛酸——HI是鹵族元素中酸性最強的無氧酸,能夠通過Brønsted酸性激活官能團[11]。碘離子不僅是較好的親核試劑和離去基團,氫碘酸(HI)還是羥基的高選擇性還原劑。由于生物質原料和其衍生的平臺化合物的多羥基特性,羥基選擇性轉化是生物質基化學品制備過程中非常重要的反應。通過化學催化實現(xiàn)生物質多羥基原料的氫解一般很難達到理想的選擇性。因此,基于碘離子和氫碘酸的獨特反應性能,HI被認為是一種理想的生物質化學轉化輔助試劑[11,12]。
近年來,HI作為還原劑用于生物質的化學轉化已有一些研究報道[13~16]。然而,HI會在加氫脫氧反應過程中不斷被氧化生成I?,導致體系中大量累積的I?很難分離和處理。為了解決這一難題,構建能夠將I?原位還原為HI并實現(xiàn)HI和I?在反應體系中的催化循環(huán)具有重要意義[17]。目前,還原碘單質為氫碘酸的方法主要有:使用紅磷、亞磷酸、次磷酸、硫化氫等作為還原劑,或者將碘蒸氣和氫氣共同通過高溫的鉑海綿等[13,18]。然而,這些方法都存在各自的問題,如反應速度慢、反應條件苛刻、會產(chǎn)生大量的污染和浪費等。針對以上HI參與生物質加氫脫氧反應所存在的難題,本文綜述了多種催化體系,包括“HI-金屬催化劑-H?”催化體系與“HI-氫供體”無金屬催化體系,可高效催化生物質及其衍生的平臺化合物轉化為高價值化學品和生物燃料,并且實現(xiàn)HI和I?的催化循環(huán)。
2 “HI-金屬催化劑-H?”催化體系
HI在生物質的加氫脫氧反應中可以發(fā)揮雙重作用:(1)提供酸性環(huán)境;(2)作為還原劑定向還原反應物,同時自身被氧化為I?。而如何在生物質HDO的同時實現(xiàn)HI與I?的催化循環(huán)至關重要。
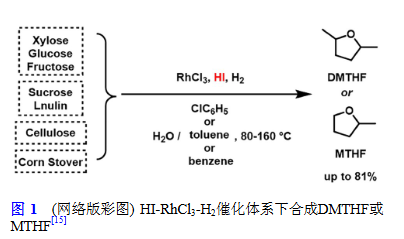
基于此,2010年,Sen等[15]報道了在“HI-RhCl?-H?”體系中將生物質衍生的碳水化合物(單糖、多糖、纖維素、木質纖維素)一步高效轉化為液體燃料2,5-二甲基四氫呋喃(DMTHF)或甲基四氫呋喃(MTHF)。其中RhCl?除了可以作為C=O和C=C鍵的加氫催化劑外,更關鍵的是其可以催化I?與H?的反應即時生成HI,使得HI繼續(xù)參與反應的催化循環(huán)[19]。在此基礎上,Hu等[20]以重金屬超富集植物(富砷蜈蚣草)為原料,在獲得高質量的生物燃料的同時實現(xiàn)了砷的回收。生物燃料總產(chǎn)量為18.8 wt%(基于蜈蚣草),生物炭中重金屬(Cu、Pb、Zn、Cd、As)含量超過50%,生物燃料中重金屬殘留量低于20%。
“HI-金屬催化劑-H?”催化體系除了在生物燃料合成方面的應用,也可進一步拓展到生物質精細化學品的合成。例如,生物質碳水化合物制備5-甲基糠醛、甘油制備丙烯、甘油酸制備3-碘丙酸、D-山梨醇制備己烯及D-葡萄糖二酸制備己二酸或3-羥基己二酸等。
5-甲基糠醛(MF)是一種重要的精細化學品,它被廣泛應用于醫(yī)藥、農(nóng)藥、化妝品、食品等行業(yè),因其具有特殊的香味,也是食品香料和煙用香精的重要組成部分,甚至被認為是重要的抗癌藥物[21]。2011年,Yang等[16]通過“HI-RuCl?”或“Pd/C-H?”催化體系,實現(xiàn)了果糖等生物質碳水化合物一步法制備MF,獲得68%的收率。并于2012年發(fā)現(xiàn)僅使用HI和水就可以獲得47%收率的MF[17],但是生成的I?需要在金屬催化劑的作用下與H?重新生成HI。此外,該反應體系也適用于甘油高選擇性合成丙烯。
然而上述體系存在貴金屬催化劑(Rh、Ru、Pd)成本昂貴等問題。基于此,2021年,Xiao等[22]發(fā)現(xiàn)低成本碳化鎢(WC)可代替貴金屬催化劑將體系中產(chǎn)生的碘單質還原為HI(WC+H?O+I?→WO?·0.33H?O+HI),但是WC在水和I?的共同作用下穩(wěn)定性差,因此通過液相沉積法對高分散負載WC進行疏水性有機硅改性,改性后的超疏水高分散負載WC@MTMS材料在果糖制備MF應用中具有較好的穩(wěn)定性,其催化活性可與貴金屬鈀碳(Pd/C)催化劑相媲美。
甘油作為生物柴油和皂化反應的副產(chǎn)物,產(chǎn)量較大但沒有合理利用,因此,由甘油制備高附加值化學品是生物質轉化領域的一個重要研究方向[23,24]。甘油的仲羥基進行氫解的選擇性較低,用化學催化的方法很難從甘油中獲得有價值的1,3-取代物。2019年,Yang等[25]通過設計將甘油換成末端含有吸電子基團的甘油酸(GA),通過氫碘酸介導的加氫反應,實現(xiàn)了溫和條件下,在3 h內定量地轉化為3-碘丙酸(3-IPA)。并提出了取代-消除-加成的反應機制,確定了該反應途徑為甘油酸→2,3-二碘丙酸→丙烯酸→3-碘丙酸。動力學研究證實了甘油酸到2,3-二碘丙酸的取代反應是決速步,且總活化能為103 kJ/mol。過程中HI作為還原劑被氧化為I?,然后在金屬催化劑和H?的作用下原位再生為HI,該催化體系穩(wěn)定,可多次重復使用而不損失活性。3-碘丙酸是一種良好的平臺化合物,可高效轉化為3-羥基丙酸、丙烯酸鈉等高附加值化學品。
“HI-金屬催化劑-H?”催化體系還可應用于生物基己烯及二元酸的合成。D-山梨醇是一種重要的生物質平臺化合物,其可以由纖維素水解后得到的葡萄糖再經(jīng)催化加氫獲得。D-山梨醇在HI和金屬協(xié)同催化下還原轉化為碘己烷,也提出了類似的反應途徑[26]。在水-環(huán)己烷兩相體系中用HI在100℃下反應3 h可高效地將D-山梨醇轉化為碘己烷(總收率94.2%,其中包括1.2%的1-碘己烷、31%的2-碘己烷和62%的3-碘己烷),并可進一步轉化為己烯(92%總產(chǎn)率,包括5%的1-己烯、61%的2-己烯和26%的3-己烯)。機理研究證實該反應遵循取代-消除-加成機理,山梨醇上的羥基從端位開始被碘離子取代,相鄰碳原子上的兩個碘發(fā)生消除反應生成C=C雙鍵,然后C=C雙鍵進一步與氫碘酸發(fā)生加成反應。經(jīng)過多次取代、消去和加成反應后生成己烯。該雙相系統(tǒng)穩(wěn)定且可重復使用。同樣,應用類似的催化體系“HI-Rh/C-H?”可以催化生物質衍生的D-葡萄糖二酸選擇性轉化為3-碘己二酸(3-IAA),3-IAA作為一種平臺化合物可進一步轉化為己二酸和3-羥基己二酸[27]。
3 “HI-氫供體”無金屬催化體系
雖然“HI-金屬催化劑-H?”催化體系展現(xiàn)了強大的催化能力,但過量的HI和金屬催化劑的使用使得該體系依舊面臨著環(huán)境、成本等諸多問題。因此,無金屬催化體系的開發(fā)具有重要意義。無金屬催化體系主要包括以下5個部分:H?PO?-HI體系、PA-HI-H?體系、H?-NaI-H?體系、H?-NaI-N?體系及FA-NaI-微波體系。
3.1 H?PO?-HI體系
1995年,Robinson[28]提出可以通過幾個催化步驟從生物質中產(chǎn)生有價值的碳氫化合物:如纖維素或淀粉首先在稀酸中水解,同時催化加氫得到山梨醇。得到的多羥基醇通過與氫碘酸和共還原劑(如磷酸或次磷酸)反應還原為液態(tài)烴和鹵代烴,同時將I?還原為HI。鹵代烴在含堿的沸騰醇溶液中發(fā)生消除反應轉化為烯烴。利用此方法使多羥基醇轉化為液態(tài)碳氫化合物的顯著特點是反應發(fā)生在相對溫和的常壓沸騰水溶液條件下,不需要昂貴的催化劑。雖然Robinson等[28]提出了從生物質到碳氫化合物這一創(chuàng)新策略的完整路線圖,但在HI還原山梨醇的過程中,反應機理并不明晰。因此,Liu等利用H?PO?-HI體系分別還原山梨醇[29]和木糖醇[13]生成碳氫化合物,他們認為碳氫化合物是直接通過碘代中間體產(chǎn)生:首先,羥基被強酸性HI可逆地質子化,生成氧鎓離子,氧鎓離子會自發(fā)地失去一個水分子,得到一個碳正離子中間體,而碳正離子中間體會與碘離子反應生成烷基碘化物,在強酸性條件下,碘的半徑較大,電負性較低,導致C−I鍵的極性反轉,因此,碳鏈上的碘原子變得親電,然后被親核的碘離子攻擊,使附著的碘原子與氫交換,從而形成I?和C−H鍵。HI可由次磷酸和I?再生。此外,使用甲酸(FA)作為化學計量還原劑,山梨醇也被還原為烷烴和烯烴的混合物[30]。
3.2 PA-HI-H?體系
HI類似于一種雙功能催化劑,具有很強的Brønsted酸性和活化H?的能力,它可以選擇性地對醚鍵和C−OH鍵進行裂解,而不攻擊高階含氧官能團,如羧酸。2017年,Vlachos等[31]和Xu等[14]利用PA-HI-H?無金屬催化體系從生物質衍生的四氫呋喃-2,5-二羧酸(THFDCA)制備得到89%產(chǎn)率的己二酸(AA),機理研究表明在HI的存在下,不需要金屬就可以促進THFDCA的氫解。在丙酸溶劑中,HI和H?協(xié)同作用,裂解THFDCA環(huán)中的C–O鍵,而在沒有H?的情況下,碘化物不能單獨驅動THFDCA的開環(huán)。THFDCA分子首先被質子化,隨后一個I?攻擊α-C,從而裂解C–O開環(huán)形成IHA,當HI和H?都存在時,IHA的C–I鍵可以氫解成HAA,HAA經(jīng)過類似途徑得到AA。其中IHA的C–I鍵氫解是決速步驟,他們在文章中推測氫分子是通過在C–I鍵激活后形成碘自由基而被活化的,從而產(chǎn)生C–H和HI,但該推測并未有強有力證據(jù)證明。有機酸溶劑可能在調節(jié)系統(tǒng)的酸堿性中起作用,促進底物質子化成氧鎓離子,從而大大加快了反應速度。2018年,Gilkey等[32]報道了一種改進的方法,其中HI被碘化鹽取代,如碘化鋰(LiI)和使用Nafion樹脂。2019年,他們[33]進一步將碘化物鹽(NaI)與沸石結合,在類似的無金屬途徑中獲得了較高的AA產(chǎn)率。
3.3 H?-NaI-H?體系
為了大幅減少酸的用量及避免貴金屬催化劑的使用,2019年,Peng等[34]進一步開發(fā)了一種H?-NaI-H?無金屬催化體系,利用I?優(yōu)異的親核取代性質和對C–O鍵斷裂的高反應活性,實現(xiàn)了無金屬催化淀粉直接制備MF。文中探討了時間、溫度、酸度、碘鹽、氫氣壓力、兩相溶劑的比例對該反應的影響。在最優(yōu)反應條件下,由淀粉可獲得38%產(chǎn)率的5-甲基糠醛和46%的總有機產(chǎn)物產(chǎn)率。通過TEMPO捕獲反應中存在的自由基中間體,用電子順磁共振波譜儀(EPR)和液相質譜儀測定了中間體的準確結構。并且通過氫氣氫解中間體模型分子2-碘甲基呋喃無需金屬參與便可得到2-甲基呋喃,再次驗證了無金屬催化。基于以上研究,提出了涉及自由基的反應機理:首先,在酸性環(huán)境中,5-羥甲基糠醛(HMF)被I?取代形成IMF,并生成一分子水。然后,IMF受熱解離成一個碘自由基和一個MF自由基。碘自由基活化了H?反應生成HI和氫自由基,氫自由基與MF自由基結合生成MF。此外,以HMF為底物,通過此方法可直接獲得80.8%產(chǎn)率的MF。
H?-NaI-H?無金屬催化體系與之前的HI-RuCl?-H?金屬催化體系的反應機理有一定的差異,造成這種差異的主要原因如下:(1)H?-NaI-H?無金屬催化體系與HI-RuCl?-H?金屬催化體系相比使用了較高的溫度(160℃ vs. 90℃),因此可以產(chǎn)生碘自由基活化氫氣;(2)H?-NaI-H?無金屬催化體系對比HI-RuCl?-H?金屬催化體系使用了較低濃度的HI(0.09 mmol·mL?¹ vs. 1.5 mmol·mL?¹),因此催化活性更高。此外,H?-NaI-H?催化體系與PA-HI-H?催化體系相比,無需添加HI,大大減少了酸的用量,且用實驗捕獲到的MF自由基證明了自由基機理。
2020年,Liu等[35]基于上述體系,提出了一種HOAc-NaI-H?的無金屬催化體系,用于生物基乳酸高選擇性轉化為丙酸,并以丙酸為溶劑簡化了分離步驟,其中反應物本身提供了反應所需的酸性,丙酸的收率最高可達99%以上。基于動力學研究和中間體捕獲實驗,他們提出了熱誘導下碘代中間體均裂產(chǎn)生自由基并進一步活化氫氣的機理途徑:首先,在乙酸(HOAc)溶劑中,LA與溶劑快速酯化,生成AOPA。然后,AOPA被I?取代,生成2-IPA,這是該反應的限速步驟。由于2-IPA不穩(wěn)定,它會迅速分裂成PA自由基和碘自由基,因此在反應過程中檢測不到2-IPA。隨后,碘自由基激活H?形成HI和氫自由基,氫自由基同時與PA自由基結合形成PA。因此,不會產(chǎn)生I?,完成催化循環(huán)。該無金屬體系可重復使用5次,活性無損失,丙酸產(chǎn)品易于分離。此外,進一步以纖維素為原料,可通過兩步法實現(xiàn)纖維素到丙酸的高效轉化。HOAc-NaI-H?催化體系是PA-HI-H?催化體系和H?-NaI-H?催化體系的結合版,無需加入HI,HOAC本身作為溶劑并提供酸性。
3.4 H?-NaI-N?體系
此前,Peng等[34]發(fā)現(xiàn)HMF在無水條件下于H?SO?-NaI-H?催化體系中可生成MF(主要產(chǎn)物)和二甲酰糠醛(DFF)(次要產(chǎn)物),水的用量的增加會導致MF的產(chǎn)量顯著增加。在此基礎上,2021年,Peng等[36]進一步開發(fā)了酸性條件下碘離子介導的醇的歧化反應體系H?SO?-NaI-N?,在180℃、N?氣氛下,以四氫呋喃(THF)為溶劑,采用NaI和硫酸可催化HMF同時制備MF和DFF,產(chǎn)率分別為49%和49%[36]。將該體系應用于其他醇類,包括具有吸電子和給電子基團的芳醇衍生物、呋喃環(huán)醇衍生物、烯丙醇衍生物和二元醇,結果表明取代基的電子效應對歧化產(chǎn)物的產(chǎn)率和比例沒有顯著影響,該反應具有優(yōu)異的官能團兼容性,且反應原子經(jīng)濟性接近100%。通過機理研究提出了一種自由基途徑,涉及碘代烷烴(用于還原)和碘醇(用于氧化)中間體:醇首先與HI反應,形成碘代物;碘代物在加熱作用下均裂成碘自由基和烷基自由基;烷基自由基與HI反應生成碳氫化合物F和碘自由基;同時,醇被碘自由基進攻形成HI和自由基D,自由基D再與另一個碘自由基結合形成碘代物G;G物質釋放一分子HI,生成目標產(chǎn)物醛H,實現(xiàn)HI-I?的相互轉化,從而完成催化循環(huán)。
3.5 FA-NaI-微波體系
根據(jù)文獻記載,I?可被甲酸還原成HI,這意味著甲酸中的氫可通過碘化物介導過程轉移到產(chǎn)物中。在此基礎上,2021年,Xu等[37]通過提高反應溫度,開發(fā)了FA-NaI-微波體系并將其應用于多種生物質催化轉化,該體系中FA作為氫供體且提供酸性。通過中間體捕獲實驗,作者提出了果糖制備MF的可能途徑:首先在甲酸催化作用下果糖脫水生成HMF,然后其羥基被碘代生成IMF,隨后經(jīng)相轉移進入有機相進而被HI還原為MF,而過程中生成的碘會被甲酸原位還原成HI。他們提出該過程可能也涉及自由基機理,因為通過自由基捕獲劑捕獲到了MF自由基,但在文章中并未重點描述。該反應體系具有普適性,糖類化合物、淀粉甚至原生生物質均可高效轉化。
4 總結與展望
氫碘酸(HI)在生物質加氫脫氧(HDO)轉化為生物基燃料或精細化學品應用中發(fā)揮重要作用,本文對HI在生物質加氫脫氧反應中的催化作用進行了系統(tǒng)的概括,即采用不同方案(金屬催化劑、提高溫度、微波等),使不同氫供體(H?PO?/H?/FA等)原位將I?還原成HI,實現(xiàn)HI和I?在反應體系中的催化循環(huán),從而實現(xiàn)了生物質的定向轉化。催化體系主要概括為“HI-金屬催化劑-H?”催化體系和“HI-氫供體”無金屬催化體系。
其中“HI-金屬催化劑-H?”催化體系大部分使用的是貴金屬,該體系展現(xiàn)出強大的催化能力,多種生物質原料均能適用。但過量的HI和金屬催化劑的使用使得該體系面臨著環(huán)境、成本等諸多問題。“HI-氫供體”無金屬催化體系包括H?PO?-HI體系、PA-HI-H?體系、H?-NaI-H?體系、H?-NaI-N?體系及FA-NaI-微波5種催化體系,從加入后會產(chǎn)生磷副產(chǎn)物的共還原劑H?PO?到H?作為還原劑,然后到N?下醇自身的氧化還原,最后到甲酸參與的氫轉移反應,“HI-氫供體”無金屬催化體系一步步發(fā)展。機理方面,一般C–OH鍵經(jīng)過I?親核取代得到C–I鍵,在H?PO?-HI體系中C–I鍵是在酸性條件下經(jīng)過極性反轉后被I?親核取代到C–H鍵,而在其他4個無金屬催化體系中,C–I鍵經(jīng)過碘自由基活化H?氫解得到C–H鍵。總體而言,無金屬體系比貴金屬體系反應成本更低、HI酸用量更少,但是反應溫度更高。
未來HI在生物質催化轉化領域的應用充滿機遇和挑戰(zhàn)。首先,需要加強機理研究,包括反應中各物質的相互作用、關鍵步驟的動態(tài)變化,以期發(fā)現(xiàn)新的催化途徑或優(yōu)化現(xiàn)有過程。目前,C–I鍵氫解為C–H鍵過程中產(chǎn)生的碘自由基并未有直接證據(jù)證實,以及IMF被認為可能是中間體,但不能排除I?直接還原的可能性等。其次,目前研究范圍主要局限在生物質的纖維素和半纖維素衍生的多元醇、糖類以及淀粉等簡單生物質原料,生物質中的木質素部分還需進一步探究,以實現(xiàn)生物質的全組分利用。最后,還需重視HI催化過程的環(huán)境影響評估,應發(fā)展更有效的回收與再利用技術,有助于減少環(huán)境污染,提高資源利用率。
參考文獻
[1] Corma A, Iborra S, Velty A. Chem Rev, 2007, 107: 2411–2502.
[2] van Putten RJ, van der Waal JC, de Jong E, Rasrendra CB, Heeres HJ, de Vries JG. Chem Rev, 2013, 113: 1499–1597.
[3] Samec JSM. Chem, 2018, 4: 1199–1200.
[4] Ingram LO, Aldrich HC, Borges ACC, Causey TB, Martinez A, Morales F, Saleh A, Underwood SA, Yomano LP, York SW, Zaldivar J, Zhou S. Biotechnol Prog, 1999, 15: 855–866.
[5] Wei H, Wang Z, Li H. Green Chem, 2022, 24: 1930–1950.
[6] Qu L, Jiang X, Zhang Z, Zhang X, Song G, Wang H, Yuan Y, Chang Y. Green Chem, 2021, 23: 9348–9376.
[7] Alonso DM, Wettstein SG, Dumesic JA. Chem Soc Rev, 2012, 41: 8075–8098.
[8] 練彩霞, 李凝, 蔣武. 化工進展, 2020, 39: 153–162.
[9] 張占輝, 劉慶彬. 化學進展, 2006, 18: 270–280.
[10] Luján-Montelongo JA, Mateus-Ruiz JB, Valdez-García RM. Eur J Org Chem, 2023, 26: e202201156.
[11] Breugst M, von der Heiden D. Chem Eur J, 2018, 24: 9187–9199.
[12] Satam JR, Jayaram RV. Catal Commun, 2008, 9: 1033–1039.
[13] Lv DC, Liu YQ, Li SR, Ye YY, Wang D. Fuel Processing Tech, 2015, 131: 325–329.
[14] Gilkey MJ, Mironenko AV, Vlachos DG, Xu B. ACS Catal, 2017, 7: 6619–6634.
[15] Yang W, Sen A. ChemSusChem, 2010, 3: 597–603.
[16] Yang W, Sen A. ChemSusChem, 2011, 4: 349–352.
[17] Yang W, Grochowski MR, Sen A. ChemSusChem, 2012, 5: 1218–1222.
[18] Gillis RJ, Lolur P, Green WH. ACS Sustain Chem Eng, 2019, 7: 7369–7377.
[19] Grochowski MR, Yang W, Sen A. Chem Eur J, 2012, 18: 12363–12371.
[20] Hu Y, Jiang H, Hu M, Liu Y, Zhao F, Yang W. Fuel, 2022, 310: 122476.
[21] Galkin KI, Ananikov VP. ChemSusChem, 2019, 12: 185–189.
[22] Xiao J, Chen Q, Yu R, Qian J, Liu Z, Li T, Yang H, Shao J, Yang W, Chen H. Appl Surf Sci, 2021, 565: 150523.
[23] Elsayed M, Eraky M, Osman AI, Wang J, Farghali M, Rashwan AK, Yacoub IH, Hanelt D, Abomohra A. Environ Chem Lett, 2024, 22: 609–634.
[24] Aprialdi F, Mujahidin D, Kadja GTM. Waste Biomass Valor, 2024, 15: 5069–5092.
[25] Li T, Liu S, Wang B, Long J, Jiang J, Jin P, Fu Y, Yu H, Yang W. Green Chem, 2019, 21: 4434–4442.
[26] Li T, Jin P, Zhang Y, Huang K, Long J, Jiang J, Liu S, Sen A, Yang W. Ind Eng Chem Res, 2020, 59: 17173–17181.
[27] Shi H, Zhang L, Wu Y, Yu R, Peng Y, Wang Y, Li T, Yang W. Catal Lett, 2020, 151: 338–343.
[28] Robinson JM. Prepr Pap-Am Chem Soc Div Fuel Chem, 1995, 40: 729–732.
[29] Lv DC, Liu YQ, Zhang BB, Wang D. Energy Fuels, 2014, 28: 3802–3807.
[30] Gunawan M, Hudaya T, Soerawidjaja TH. J Eng Technol Sci, 2021, 53: 210106.
[31] Fu J, Vasiliadou ES, Goulas KA, Saha B, Vlachos DG. Catal Sci Technol, 2017, 7: 4944–4954.
[32] Gilkey MJ, Balakumar R, Vlachos DG, Xu B. Catal Sci Technol, 2018, 8: 2661–2671.
[33] Gilkey MJ, Cho HJ, Murphy BM, Wu J, Vlachos DG, Xu B. ACS Appl Energy Mater, 2020, 3: 99–105.
[34] Peng Y, Li X, Gao T, Li T, Yang W. Green Chem, 2019, 21: 4169–4177.
[35] Liu S, Feng H, Li T, Wang Y, Rong N, Yang W. Green Chem, 2020, 22: 7468–7475.
[36] Peng Y, Huang Y, Li T, Rong N, Jiang H, Shi H, Yang W. Green Chem, 2021, 23: 8108–8115.
[37] Xu J, Miao X, Liu L, Wang Y, Yang W. ChemSusChem, 2021, 14: 5311–5319.




