摘要:摘要:利用攪拌摩擦焊技術連接低熔點的銅與鋁�,連接過程中銅原子與鋁原子在界面的擴散能力影響界面力學性能。采用分子動力學研究攪拌摩擦焊過程中銅/鋁界面原子的擴散行為���,評價了不
摘要:利用攪拌摩擦焊技術連接低熔點的銅與鋁�����,連接過程中銅原子與鋁原子在界面的擴散能力影響界面力學性能。采用分子動力學研究攪拌摩擦焊過程中銅/鋁界面原子的擴散行為����,評價了不同應變速率下界面的力學性能����。結果表明:銅原子擴散進入鋁晶格的數量遠大于鋁原子擴散進入銅晶格的數量�,原子擴散以最近鄰跳躍機制為主,銅原子和鋁原子的擴散激活能分別為0.58�����、0.75eV����。界面過渡層厚度主要受保溫溫度控制,800K下的界面為最佳厚度界面;拉伸應變速率為 (1×10^{10}s^{-1}) 時,界面抗拉強度最大,達到3.19GPa。塑性變形在鋁側�,而銅側幾乎沒有塑性變形���,變形過程中發生的位錯反應由全位錯分解和壓桿位錯合成�����,位錯能下降��,是以肖克萊位錯為主要的滑移位錯。
關鍵詞:分子動力學;Cu/Al界面;擴散;應變速率;力學性能
論文《銅_鋁攪拌摩擦焊接頭界面原子擴散及力學性能模擬》發表在《蘭州理工大學學報》��,版權歸《蘭州理工大學學報》所有��。本文來自網絡平臺����,僅供參考��。
引言
由于銅具有優異的導電性和導熱性以及鋁的低比重量和低成本,銅/鋁異種接頭廣泛應用于工業領域��,如作為電連接器���、熱交換管�����、制冷管等����,因此銅鋁的連接技術成為影響接頭性能的關鍵���。
攪拌摩擦焊(FSW)作為一種固態焊接技術連接異種金屬材料��,可以避免金屬連接過程中因發生熔化而出現氧化物�����、金屬夾雜等缺陷,達到提高接頭強度的目的��。然而�����,攪拌連接過程中各工藝參數匹配不佳時會同樣導致接頭界面產生隧道��、孔洞、微裂紋等缺陷��,局部弱連接的產生往往是界面上原子的不充分擴散導致的����,因此研究焊接過程中原子的擴散行為���,是獲得性能優異接頭的有效途徑����。
分子動力學(MD)是研究固態連接時原子擴散以及金屬間化合物形成有效的方法。羅龍等模擬了相同保溫時間不同保溫溫度下銅/鋁界面原子的擴散過程���,得出銅原子向鋁側擴散的結論。Chen等固定保溫時間為0.2ns����,模擬了不同溫度及壓力下的Cu-Ag擴散連接�,得出擴散過渡層厚度與壓力有關���,在高溫時界面過渡區域為非晶態��,冷卻到室溫時又從非晶態轉變為晶態結構�����。Li等固定保溫時間為6ns,模擬了Cu-Al界面原子擴散的機制����,得出Cu在Al點陣中的擴散機制遵循最近鄰跳躍機制����。Mypati等在MD模擬中,根據試驗測得的峰值溫度,發現在498℃下�,Al在Cu晶格中擴散形成了(gamma-Cu_9Al_4)金屬間化合物���。Li等采用分子動力學模擬方法研究壓力對Cu/Al異種攪拌摩擦焊界面擴散行為的影響,得出壓力對擴散層厚度的影響較小����。Liu等利用分子動力學模擬研究Ti/Al界面的擴散行為和力學性能����,得出隨溫度升高���,主要發生Ti原子向Al原子擴散���,導致界面原子的晶格畸變和擴散層厚度增加�,拉伸結果表明,斷裂主要發生在鋁側。盡管進行了如此多銅/鋁界面的分子動力學模擬,但對保溫溫度��、保溫時間等影響因素的綜合分析仍然較少�。
本文研究不同保溫溫度和保溫時間對Cu-Al界面原子擴散行為的影響,并在300K下研究了擴散界面在不同應變速率下的力學性能�。通過徑向分布函數(RDF)�����、均方位移(MSD)、原子濃度變化趨勢�����、能量���、體積���、過渡層厚���、抗拉強度和屈服強度等全面分析了Cu/Al界面的擴散行為和力學性能�����。
1 模擬方法
利用開源分子動力學軟件Lammps對銅/鋁界面層的擴散和力學性能進行了模擬研究。通過開放式可視化工具(Ovito)對所有計算結果進行后處理和可視化��。通過Ovito的鍵角分析和位錯分析計算了晶格結構和缺陷分布�。借助嵌入原子方法勢(EAM)定義Al和Cu的原子間相互作用,EAM中體系原子的總勢能為:
[E_{total}=sum_{i} F_{i}(
ho_{i})+frac{1}{2}sum_{j
eq i} phi_{ij}(r_{ij}) quad(1)]
其中:(F_{i}(
ho_{i}))為原子的嵌入能;(phi_{ij}(r_{ij}))為原子i和j之間的對勢;(r_{ij})為原子i和j之間的相對位移;(
ho_{i})為原子i和其他原子產生的背景電子密度。
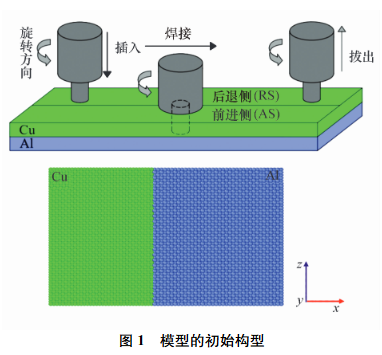
焊接過程為攪拌頭插入�����、焊接�、拔出。MD模型大小為(138.41×10^{-12}m×101.22×10^{-12}m×101.22×10^{-12}m)�,擴散過程中x方向采用收縮邊界條件����,y��、z方向采用周期性邊界條件����。單晶銅板與鋁板的接觸面為(100)����,銅、鋁原子數分別為53312和47500���,沿x方向在銅板和鋁板的外圍分別固定3層原子作為模型邊界。分別在650�����、700���、750���、800�、850K下進行銅鋁界面擴散過程模擬。所有原子的初始速度均符合麥克斯韋分布。采用Velocity-Verlet方法對原子的牛頓運動方程進行數值積分,時間步長為1fs�����。該結構首先在1K下弛豫23ps�����,然后在21ps內加熱至保溫溫度����。隨后�,采用Nose/Hoover熱浴法使溫度保持恒定,保溫時間為1000ps����。加熱和保溫過程采用恒定原子數�����、恒定壓力、恒定溫度(NPT)系綜,保持外部壓力處于大氣壓值�。
選取保溫溫度為800K來研究擴散界面的力學性能����,并在300K不同應變速率下進行單軸拉伸��,應變速率分別為(5×10^8s^{-1})���、(1×10^9s^{-1})�、(5×10^9s^{-1})�����、(1×10^{10}s^{-1})。
2 結果分析
2.1 界面原子的擴散
不同溫度下保溫1000ps后,Al原子和Cu原子都穿過了初始界面��,擴散進入了對方點陣結構中�,在界面形成了銅-鋁復合層。過渡層厚度、原子擴散進入對方晶格點陣中的距離都隨保溫溫度升高而改變。鋁原子、銅原子各自在對方晶格點陣中的擴散能力有明顯的差異��。溫度低于700K時����,鋁原子、銅原子向對方點陣中的擴散不明顯�����。溫度升高到750K����,銅原子擴散進入鋁晶格的數量增多,離原始界面的距離增大���。提高保溫溫度到850K,銅原子的擴散更顯著�����。比較銅原子����、鋁原子越過初始界面的原子數量、離開原始界面的距離�����,認為界面發生的是Cu原子向Al晶格中的單向擴散�,Al原子只在界面處擴散��。
800K下不同保溫時間的擴散狀態顯示���,仍以原始界面為基準線��,保溫200ps時���,銅原子明顯向鋁原子一側擴散�����。保溫時間延長,銅原子向鋁原子晶格中擴散得更顯著����,表現在銅原子越過初始界面的數量增多���,擴散進入鋁晶格中的距離增大(銅原子距離原始界面的最遠距離)����。當保溫時間達到1000ps���,銅原子在鋁晶格中的分布均勻�。相較而言,隨時間延長��,鋁原子擴散的數量��、穿過界面的距離并未發生明顯變化���,仍在界面處擴散��。
產生這種結果的原因有3點:首先�,銅的熔點為1356K,鋁的熔點為933K�,銅的熔點比鋁高423K�,相同溫度下鋁晶格中更容易產生空位和缺陷�,有利于銅原子向鋁晶格中擴散;其次,金屬的熔點越高����,金屬鍵越強��,越難斷裂,使得Al原子很難擴散到Cu晶格中;第三�����,鋁原子半徑(0.143nm)大于Cu原子半徑(0.128nm)�����,原子半徑小的銅原子更容易擴散到原子半徑大的鋁晶格中���。
2.2 擴散系數和過渡層厚變化
溫度升高�����,Cu、Al原子均方位移(MSD)增大。因為溫度為原子擴散激活能的指數函數,為原子擴散提供了動能�,實驗僅研究原子在x方向的一維擴散�,其中N是系統的維度�,視為1。在此基礎上,根據愛因斯坦擴散定律���,擴散系數為:
[D=lim_{t o infty} frac{1}{2t}left<|r_z(t)-r_z(0)|^2
ight> quad(2)]
均方位移:
[L_{MSD}=left=frac{1}{N}sum_{i=1}^{N}left(|r_{zi}(t)-r_{zi}(0)|^2
ight) quad(3)]
其中(r_{zi}(t))為原子在t時刻的位置,(r_{zi}(0))為原子的初始位置�,N是體系的原子數���。
擴散系數計算結果顯示,溫度低于750K時,Al原子的擴散系數隨溫度略有增大��,當溫度升高到800K時�����,鋁原子的擴散系數急劇增大�。表明溫度達到800K時�����,空位濃度急劇增大�����,鋁原子更容易越過勢壘�����,擴散驅動力增大。均方位移隨溫度、時間變化都表現出了相同的趨勢。這些模擬的結果與擴散界面實際情況相契合�����,原因在于同一溫度下銅原子間鍵能高于鋁原子間鍵能����,鋁原子鍵容易斷裂,為銅原子擴散進入鋁點陣提供了有利條件�����。根據阿倫尼烏斯公式(Arrhenius)���,得到Cu原子和Al原子的擴散激活能分別為0.58�、0.75eV�����,與相關文獻計算得出的0.50��、0.77eV接近。結合面為(100)面����、晶格發生畸變�、原子活躍度提高等因素都有利于空位的產生和界面附近原子的擴散��。
標準大氣壓條件下��,分別加熱到650、700����、750�、800�����、850K下保溫1000ps后的徑向分布函數(g(r))用來描述體系結構的有序性:
[g(r)=frac{dN}{4pi r^2
ho dr} quad(4)]
對比不同溫度下的徑向分布函數發現:隨著溫度升高���,銅�、鋁原子均呈現峰寬增加�、峰高降低的趨勢,但相同條件下���,鋁原子峰值低于銅原子,且隨溫度升高鋁原子峰數量明顯減少����,銅原子峰的數量并未隨溫度升高而減少。表明鋁原子側無序化明顯�����,這種變化趨勢歸因于溫度升高��,原子熱運動加劇�。
不同溫度下體系總能量和體積的變化顯示�����,高于800K時�����,能量和體積的變化趨勢很明顯��,這是溫度高導致原子擴散速度增大的原因;而低于750K下,能量和體積在固定值附近先波動后穩定下來�����,這表明晶格轉變已經完成�����。相變曲線全過程分為3個階段����,第一階段(0~21ps)為升溫過程��,FCC結構快速轉換����,少量轉換為BCC��、HCP����,大量轉換為無序結構;第二階段(21~200ps)為保溫過程�����,因溫度不同出現了不同的現象���,低于750K時��,從21ps開始結構轉變已經達到穩定化,而高于800K時��,結構轉變仍在繼續;第三階段(200~1021ps)仍屬于保溫階段,所有溫度下相結構轉變都達到穩定����,不再發生改變�����。這是由于溫度越高,Al-Al鍵斷裂越多����,無序結構比例越高�,Al側完全無序化轉變所需時間越短���。
分子動力學模擬中一般將擴散原子濃度均超過5%的區域定義為過渡層�����。隨溫度升高,原子在x方向擴散的距離越遠��。1000ps下650���、700����、750、800��、850K的過渡層厚度分別為(7.864×10^{-4}mu m)�、(8.026×10^{-4}mu m)、(8.943×10^{-4}mu m)、(11.816×10^{-4}mu m)、(13.201×10^{-4}mu m)��,表明溫度越高��,過渡層越厚�,且在800K下急劇加厚��。這是由于溫度較低時原子運動速度較慢����,溫度升高后參與擴散的原子數量增加,一方面使得過渡層厚度加厚,另一方面為生成金屬間化合物提供了條件,最終影響銅/鋁界面的力學性能��。比較850K�、800K下的過渡層厚度,800K時增長減緩,這是因為升溫到800K時�����,金屬原子的擴散速率增加�����,但與此同時,擴散過程中的相互碰撞也會增加�,使得擴散的速度減慢����,最終達到一個動態平衡狀態��,擴散速率接近平衡����,與Mypati等提出的銅鋁界面最佳均勻化溫度為783K的實驗結果基本一致����。因此,選擇保溫溫度800K來研究保溫時間對銅/鋁界面擴散的影響�����。
800K時不同保溫時間下�,銅、鋁原子沿著x方向的濃度分布和過渡層厚度顯示��,200�、400、600��、800�、1000ps的過渡層厚度依次為(11.546×10^{-4}mu m)、(11.683×10^{-4}mu m)�����、(11.694×10^{-4}mu m)����、(11.739×10^{-4}mu m)、(11.816×10^{-4}mu m)���,即隨保溫時間延長��,過渡層厚度逐漸增厚��,但增厚的趨勢并非線性關系,時間短于1000ps時,過渡層厚增長緩慢。然而,如果保溫溫度太低���,因擴散速率低,即使延長保溫時間��,過渡層厚度仍然較薄�����。比較保溫溫度和保溫時間對過渡層厚度增長的影響�����,顯然時間的影響弱于溫度。
2.3 擴散機理
為了驗證銅原子在鋁中的擴散機制�,在假設所有鋁原子都停留在晶格位置的基礎上�,觀察單個銅原子的運動軌跡發現��,銅�����、鋁原子擴散存在2種擴散機制:空位擴散機制和間隙擴散機制?�?瘴粩U散機制按2種途徑擴散:一種是最近鄰跳躍機制;另一種是次近鄰跳躍機制����,銅原子占據的是鋁原子的晶格結點位置�。間隙跳躍機制中���,銅原子占據鋁晶格點陣的間隙�,即八面體間隙位置���,通過躍遷實現轉移�。
統計0~100ps內銅原子在鋁側的跳躍頻率,最近鄰跳躍機制�����、次近鄰跳躍機制、間隙跳躍機制的跳躍頻率分別為14、2��、1次,即最近鄰跳躍機制占比為82.4%�,次近鄰跳躍機制占比11.8%,間隙跳躍機制占比5.9%����。顯然擴散機制以最近鄰跳躍機制為主����。
2.4 力學性能分析
攪拌摩擦焊接頭拉伸斷裂試樣顯示,斷裂發生在鋁側。650、700��、750�、800、850K不同溫度下擴散界面經300K弛豫后,在(1×10^{10}s^{-1})拉伸應變速率下的單軸拉伸應力-應變曲線顯示�,對應的抗拉強度分別為3.25�����、3.22、3.24、3.19��、3.02GPa����。850K時擴散界面抗拉強度最低,且沒有屈服現象發生。850K擴散界面隨應變率增大時��,界面的微觀結構演化顯示���,在應變為0.1時����,鋁側出現了微裂紋;應變增大到0.15時���,微裂紋擴展�,成為貫穿空洞;應變繼續增大到0.2時,空洞擴展�����、尺寸增大�,最終導致鋁側斷裂,與銅/鋁攪拌摩擦接頭單軸拉伸實驗的斷裂位置一致�����。
800K擴散界面在300K溫度下不同拉伸應變速率的實驗結果顯示�����,隨拉伸應變速率的增加�,界面的抗拉強度�、屈服強度都增大。A�、B���、C��、D各點對應的抗拉強度分別為3.19、3.07��、2.89���、2.51GPa����,E、F�����、G�����、H為屈服點�����,強度分別為2.87、2.59�、1.99���、1.85GPa����。
800K保溫后界面在300K以(5×10^9s^{-1})應變速率拉伸過程中的微觀結構演變顯示�����,因原子的無序排列�,層錯在Al側形核�����,增大應變后層錯比例提高���,最終貫穿鋁側區域,成為界面發生斷裂的原因之一���。塑性變形過程中參與滑移的位錯,主要位錯的Burgers矢量為(b=1/6〈112〉)��。位錯滑移遇到銅/鋁界面時�����,因該界面的勢壘很高����,未穿越銅/鋁界面�,Cu側沒有發生塑性變形。
在拉伸變形過程中,首先發生1個全位錯分解為2個肖克萊位錯��,2個肖克萊不全位錯夾著一個層錯區�����,層錯區的邊界線就是肖克萊位錯���,位錯能下降1/6��。此外,還發生了2個肖克萊位錯合成壓桿位錯的反應,位錯能下降5/18��。
隨應變的增大�����,鋁側無序原子結構和密排六方結構比例增大��,缺陷增多。這些缺陷和無序結構的產生是由于銅原子向鋁原子擴散過程中發生了柯肯達爾效應�����,在Al側界面產生了空洞���。
為跟蹤拉伸變形過程中位錯的演化�����,利用DXA方法計算位錯長度發現��,灰色區域為界面發生塑性變形(從屈服點開始至應力達到最大抗拉強度)的階段,在此過程中各類型位錯線變化與拉伸應變速率有關。應變速率較低時����,位錯線總長度先縮短后增長;全位錯的數量(Burgers矢量為1/2〈110〉)幾乎沒有變化;柏氏矢量為1/6〈112〉的肖克萊(Shockley)不全位錯數量不斷增多且遠大于其他類型的位錯數量�����,因此推斷肖克萊位錯在銅/鋁界面的塑性變形過程中起主導作用。應變速率較高時,位錯線總長度呈增長趨勢;全位錯的數量在屈服點之前就開始有減小��,原因是位錯發生分解所致��。
3 結論
采用分子動力學模擬了不同溫度和時間條件下Cu�、Al原子在Cu/Al界面的擴散過程及界面在300K下不同應變速率下的拉伸過程�����,得到如下結論:
1) 界面主要是銅原子向鋁原子的單向擴散�,擴散機制為最近鄰擴散機制�。從650K開始,隨保溫溫度升高,鋁側原子無序化程度提高�����。800K保溫時�����,時間低于1000ps過渡層厚度增長緩慢。
2) 界面在300K下拉伸����,850K保溫時擴散界面的抗拉強度明顯下降���,且無屈服���,拉伸過程中鋁側產生空洞�,隨空洞不斷合并���、長大����,最終撕裂;保溫溫度800K的界面結合最佳;微觀結構分析表明,層錯在Al側形核����、并逐步充滿整個區域�,導致界面區域受到嚴重破壞;而銅側未發生塑性變形��。
3) 拉伸變形過程中�,在(varepsilon=0�、0.08���、0.11)時鋁側產生大量位錯�,位錯反應有全位錯的分解和壓桿位錯的合成�����。在塑性變形過程中,Shockley型位錯的數量遠大于其他類型位錯的數量�,起主導作用���。在變形過程中�,隨應變速率增大�����,Shockley位錯增長速度減慢����。
參考文獻
[1] 何凱,胡德安�����,陳益平���,等. 鋁/銅異種材料激光焊焊縫形成機制[J]. 南昌航空大學學報(自然科學版)�,2020,34(1):7-11.
[2] 龐嘉堯�����,楊宏���,程偉���,等. 銅-鋁合金攪拌摩擦焊研究進展[J]. 金屬加工(熱加工)�,2021(2):53-59.
[3] 余章欽,胡建華���,楊正�,等. 銅/鋁磁脈沖半固態輔助釬焊界面擴散過程[J]. 焊接學報,2023,44(4):120-128,136.
[4] 金玉花�,周子正���,邢逸初�,等. 滾動軋制對7050鋁合金FSW接頭組織的影響[J]. 蘭州理工大學學報��,2023��,49(4):30-34.
[5] XU XL,LIU QY,QI S�,et al. Effect of incomplete penetration defects on mechanical and fatigue properties of friction-stir-welded 6802-T6 joint[J]. Journal of Materials Research and Technology����,2021��,15:4021-4031.
[6] MENG XC�,HUANG YX����,CAO J,et al. Recent progress on control strategies for inherent issues in friction stir welding[J]. Progress in Materials Science�,2021�����,115:100706.
[7] 韓雪杰,郭巧能�,楊仕娥����,等. 溫度和保溫時間對銅/鋁薄膜的界面擴散性能及力學性能影響的分子動力學模擬[J]. 熱加工工藝�,2019,48(14):102-107.
[8] DAS A,DE ZHI L,DAVID W,et al. Joining technologies for automotive battery systems manufacturing[J]. World Electric Vehicle Journal�����,2018�,9(2):22.
[9] DAS A���,LI D���,WILLIAMS D�,et al. Weldability and shear strength feasibility study for automotive electric vehicle battery tab interconnects[J]. Journal of the Brazilian Society of Mechanical Sciences and Engineering,2019���,41(1):54.
[10] SUN BQ,LU LX,ZHU Y. Molecular dynamics simulation on the diffusion of flavor,O? and H?O molecules in LDPE film[J]. Materials,2019�,12(21):3515.
[11] 羅龍���,王寶峰�,李麗榮. 銅/鋁熱軋擴散復合界面擴散的分子動力學模擬[J]. 熱處理技術與裝備�,2011,32(2):55-60.
[12] CHEN SD,SOHAK�����,KE FJ. Molecular dynamics modeling of diffusion bonding[J]. Scripta Materialia,2005,52(11):1135-1140.
[13] LI C,LI DX���,TAO XM,et al. Molecular dynamics simulation of diffusion bonding of Al-Cu interface[J]. Modelling and Simulation in Materials Science and Engineering,2014���,22(6):065013.
[14] MYPATIA O,KUMAR BPP,MD API,et al. Molecular dynamics simulation of atomic diffusion in friction stir spot welded Al to Cu joints[J]. Mechanics of Advanced Materials and Structures��,2021����,29(27):6053-6059.
[15] LI QH,DONG ZB,JI SD,et al. Effects of welding parameter on atom-scale interfacial diffusion behavior of Al/Cu dissimilar friction stir welding[J]. Physica Status Solidi,2021,258(10):2100123.
[16] LIU JY����,ZHANG LQ. Investigation on the diffusion behaviors and mechanical properties of the Ti/Al interface using molecular dynamics simulation[J]. Journal of Materials Engineering and Performance����,2024��,33(6):2920-2939.
[17] PLIMPTON S. Fast parallel algorithms for short-range molecular dynamics[J]. Journal of Computational Physics,1995����,117(1):1-19.
[18] STUKOWSKI A. Extracting dislocations and non-dislocation crystal defects from atomistic simulation data[J]. Modelling and Simulation in Materials Science and Engineering��,2010,18(8):015012.
[19] ACKLAND GJ��,JONES AP. Applications of local crystal structure measures in experiment and simulation[J]. Phys Rev B�,2006,73(5):054104.
[20] CAI J��,YE Y. Simple analytical embedded-atom-potential model including a long-range force for fcc metals and their alloys[J]. Phys Rev B��,1996����,54(12):8398-8410.
[21] 林雪�����,于美杰����,王成國��,等. 原子徑向分布函數在材料結構研究中的應用[J]. 材料導報�,2012�����,26(3):116-119.
[22] TRATH��,OKAZAKI M,HAO DD. Tensile fracture behavior of Cu/Al butt friction stir welding:role of interface morphology[J]. Journal of Materials Engineering and Performance,2022����,31(2):1039-1045.




